+86(311)6656 8233
 服务内容
服务内容1.DMF/VMF文件编写:为原料药、中间体、包材、辅料等产品生产企业提供药物主文件申请服务。
2.审评支持:缺陷答复(直至通过FDA审评)、DMF/VMF增补(行政和/或质量变更)、年度报告等服务。
3.美国代理服务:专业合规代理,确保沟通渠道畅通。
如需定制化方案,欢迎联系我们获取专业支持。
注册介绍
1.定义与背景
DMF(药物主文件)是美国食品药品监督管理局(FDA)要求的一种非公开技术文件,由药品原料、辅料、包装材料或生产工艺的供应商提交,用于支持新药申请(NDA)、仿制药申请(ANDA)等注册流程。其核心目的是保护供应商的技术机密,同时允许制剂厂商在申报时直接引用DMF备案号,简化审查流程,缩短药品上市时间。
VMF(兽药主文件):与DMF类似,但适用于兽药领域,提交要求与流程基本一致。
2.注册适用范围
美国DMF分为4类:
Type II:原料药、原料药中间体、制备时所用的原材料或制剂。
Type III:包装材料。
Type IV:辅料,着色剂、香料及其原料。
Type V:FDA可接受的参考信息。
3.注册基本要求
文件完整性:需提供生产工艺、质量控制、稳定性研究、分析方法验证等数据,按ICH M4的CTD格式编写(电子提交需符合eCTD标准)。
美国代理:非美国企业强烈建议指定美国代理,负责与FDA沟通及文件接收。
合规承诺:承诺符合cGMP(现行药品生产质量管理规范)并定期更新文件(至少每年提交一次年报);重大变更需及时提交增补文件。
4.注册流程
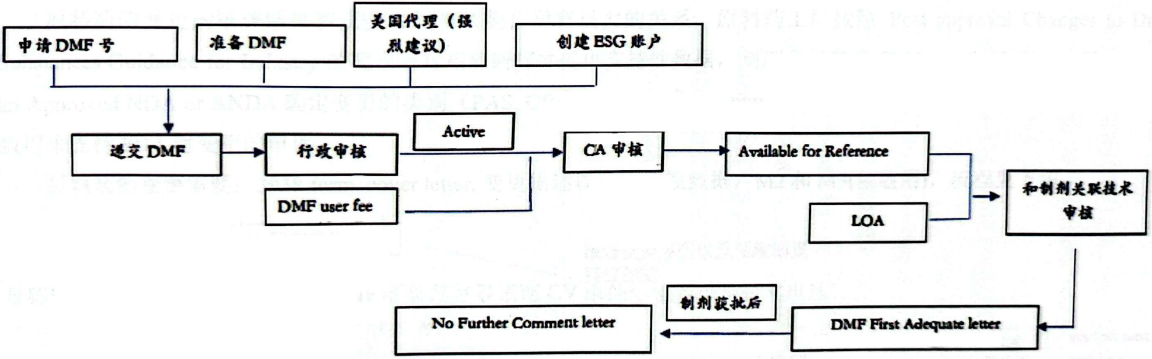
备注:如果没有制剂工厂引用,DMF缴费后只会完成CA(Completeness Assessment完整性审核)审评,不会进行技术审评。
5.法规依据
21 CFR 314.420:DMF提交与引用的法律框架。
ICH Q7:原料药GMP标准,DMF持有人需承诺符合该规范。
GDUFA(仿制药使用者付费法案):针对II型DMF的完整性审评与缴费要求。
FDA指南文件:
《Guidance for Industry: Drug Master Files》
《Container Closure Systems for Packaging Human Drugs and Biologics》(针对Type III DMF)



在线咨询Message Advisory

