+86(311)6656 8233
 服务内容
服务内容1. 法规策略咨询:协助选择最优审评程序(如CP/DCP/MRP),评估注册路径与成本。
2. 注册资料准备与审核:编制CTD文件(药学、非临床、临床模块),确保符合EMA或成员国要求。
3. MAH代理与资质申请:提供欧盟境内MAH代理服务,协助完成MIA申请及GMP合规整改。
4. 审评沟通与现场检查支持:协调与EMA或成员国药监机构的沟通,辅导应对QP检查及GMP现场审计。
5. 生命周期管理:上市后变更备案、PSUR(定期安全性更新报告)提交及证书续期服务。
如需定制化方案,欢迎联系我们获取专业支持。
注册介绍1. 定义与背景
欧盟药品制剂注册是指通过欧洲药品管理局(EMA)或成员国药监机构的审评程序,使药品在欧盟市场合法上市的过程。欧盟作为全球重要的药品监管市场,其注册体系以严格的质量、安全性和有效性要求著称。对于中国药企而言,进入欧盟市场不仅是国际化战略的关键一步,更是提升生产质量体系和全球竞争力的重要途径。
2. 注册适用范围
新药:包括创新化学药、生物制品(如单抗、重组蛋白)及孤儿药(罕见病药物)。
仿制药:需通过证明与原研药生物等效性,适用非集中审批程序(DCP)或互认程序(MRP)。
特定类别药品:如植物药、改良型药物(如缓控释制剂)及已在欧盟成员国上市的药品扩展市场。
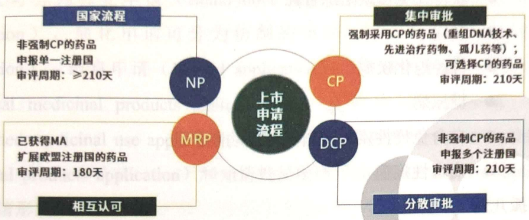
3. 注册基本要求
资质要求:
MAH(欧盟药品上市许可持有人):必须是欧盟境内的注册公司。
MIAH(制剂进口商):必须有欧盟GMP证书等。
质量管理体系、人员资质(如QP质量受权人)、设施合规性等。
技术文件:需提交CTD格式的完整资料,涵盖药学、非临床及临床数据(如适用)。
质量体系:符合EU GMP附录要求,包括工艺验证、稳定性研究及杂质控制(如致突变杂质需符合ICH M7)。
4. 注册流程
以分散审评(DCP)为例
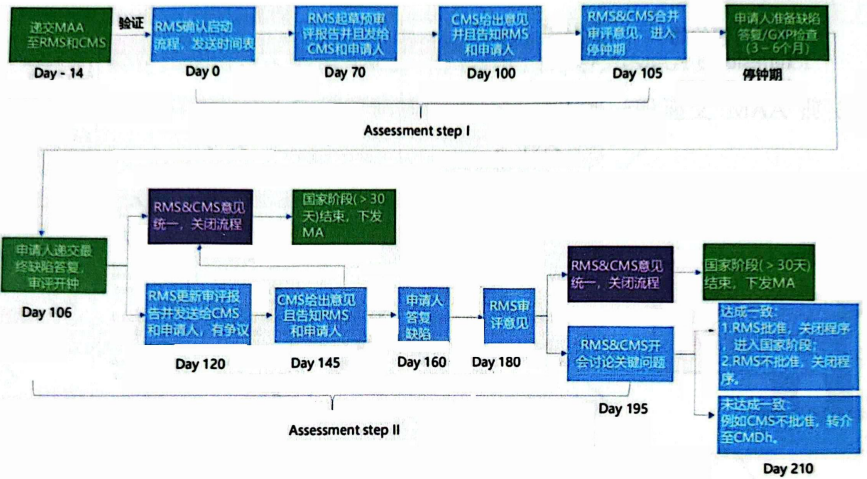
DCP程序递交流程:
预约递交(确定RMS,预约timeslot)→ Pre-submission(递交前至少2个月,可选) → 递交MAA(通过CESP或者National portal)→验证(Technical validation + regulatory validation)→缴费 (MAA递交之前(proof of payment))/之后缴费。
DCP程序审批流程:
5. 法规依据
Directive 2001/83/EC:规范人用药品的上市许可、生产与销售。
EU GMP指南:涵盖生产质量管理体系的具体要求。
ICH指导原则:如ICH Q11(原料药开发)、ICH M7(致突变杂质控制)等。
特殊产品法规:如生物制品遵循EMA生物类似药指南,植物药参考HMPC标准。



在线咨询Message Advisory

