+86(311)6656 8233
 服务内容
服务内容1. 差距分析与预审:评估企业软硬件与ASMF要求的符合性。
2. 文件编制与递交:撰写CTD/Ectd/NeeS文件,包括M1-M3模块。
3. 生命周期管理:协助处理缺陷及变更(如年缺陷答复、年度报告、主要变更)等。
4. 法规动态跟踪:实时更新欧盟及EDQM政策变化,规避合规风险。
如需定制化方案,欢迎联系我们获取专业支持。
注册介绍1. 定义与背景
活性物质主文件(ASMF),以前称为欧洲药物主文件(EDMF),是由原料药生产商向欧洲药品管理局(EMA)或欧盟各成员国药政递交的包含原料药保密信息的技术文件,用于支持制剂生产商的上市许可(MA)申请。
2. 注册适用范围
ASMF使用于以下三类原料药的申请:
新的活性物质;
已存在的活性物质,但还未被欧洲药典或欧盟成员国的药典收录;
已被欧洲药典或欧盟成员国的药典收录的药物活性物质。
3. 注册基本要求
ASMF申报资料中的M2和M3分为公开部分(AP)和保密部分(RP)。
ASMF的申报必须与使用该原料药的制剂MA申请进行关联(不早于MA一个月和不晚于MA递交),原料药的生产商通过开具授权信(LOA)实现与制剂上市许可申请的关联审评,即ASMF的审评程序和相应的制剂上市许可申请的审评程序是一致的。
4. 注册流程
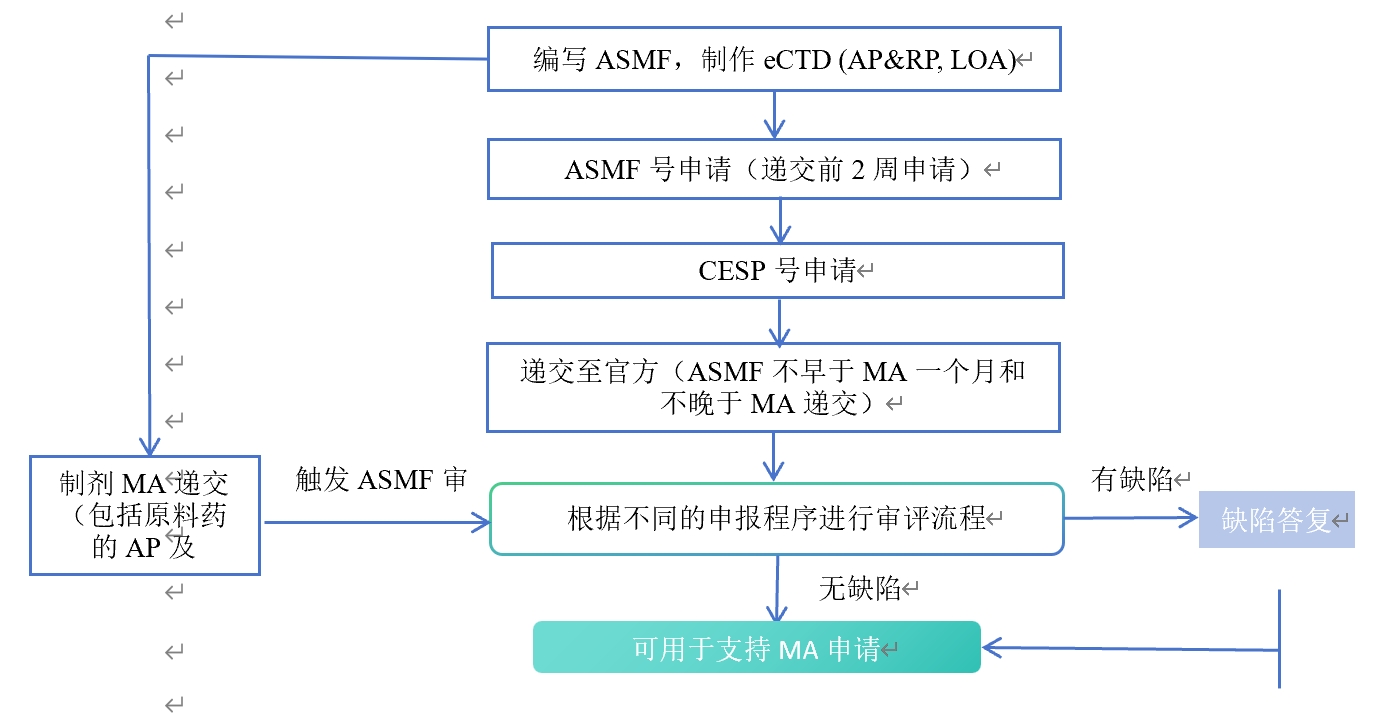
5. 法规依据
欧盟指南<Guideline on Active Substance Master File Procedure>



在线咨询Message Advisory

