+86(311)6656 8233
Services > Regulatory Registration > International Registration Services > ASMF/EDMF Registration (EU)
ASMF/EDMF Registration (EU)
 Our Services
Our Services1. Gap Analysis & Pre-assessment: Evaluate company's hardware/software compliance with ASMF requirements.
2. Documentation Preparation & Submission: Prepare CTD/eCTD/NeeS files including Modules M1-M3.
3. Lifecycle Management: Assist with deficiency responses and variations (e.g., annual deficiency responses, annual reports, major changes).
4. Regulatory Monitoring: Track real-time updates on EU/EDQM policy changes to mitigate compliance risks.
For customized solutions, please contact us for professional support.
Registration Introduction1. Definition and Background
The Active Substance Master File (ASMF), formerly known as the European Drug Master File (EDMF), is a confidential technical document submitted by active pharmaceutical ingredient (API) manufacturers to the European Medicines Agency (EMA) or EU national competent authorities. It supports the Marketing Authorization (MA) application of finished drug product manufacturers.
2. Applicable Registration Scope
ASMF applies to the following three categories of APIs:
·New active substances
·Existing active substances not yet included in the European Pharmacopoeia or EU national pharmacopoeias;
·Active substances already included in the European Pharmacopoeia or EU national pharmacopoeias.
3. Basic Registration Requirements
·The M2 and M3 sections of ASMF documentation are divided into an Applicant's Part (AP) and a Restricted Part (RP).
·ASMF submission must be linked to the MA application of the finished product using the API (not earlier than one month before and not later than the MA submission).
·The API manufacturer provides a Letter of Access (LOA) to enable joint review with the MA application. The ASMF review process aligns with the corresponding MA application review process.
4. Registration Process
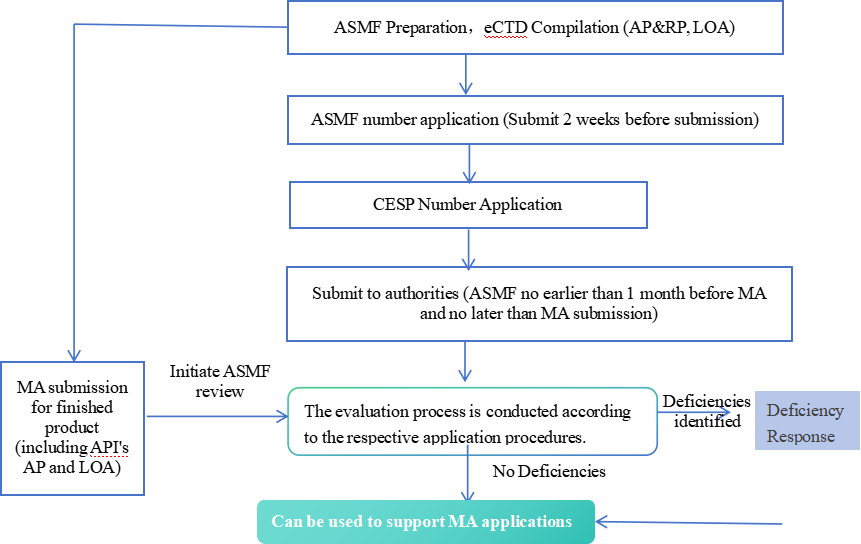
5. Regulatory Basis
EU Guideline
Product RecommendationLearn more about related businesses


Message Advisory
 Official account
Official account WeCom
WeCom- +86 (311) 6656 8233
- info@evalaboratories.com
- Chinese company address10th Floor, Building 1, Tech Centre, No.856, Zhongshan East Road, High-Tech Zone, Shijiazhuang, Hebei
- US company address813 Towne Center Drive, Pomona, CA 91767
©2025 Shijiazhuang Elab Biotech Co., Ltd. All Rights Reserved
冀ICP备2025127575号-1
冀ICP备2025127575号-1