+86(311)6656 8233
Services > Regulatory Registration > International Registration Services > European Drug Product Registration
European Drug Product Registration
 Our Services
Our Services1. Regulatory Strategy Consultation: Assist in selecting optimal review procedures (e.g., CP/DCP/MRP) and evaluating registration pathways and costs.
2. Registration Dossier Preparation & Review: Compile CTD documents (quality, non-clinical, clinical modules) to ensure compliance with EMA or national requirements.
3. MAH Representation & License Application: Provide EU-based MAH representation services and assist with MIA applications and GMP compliance rectification.
4. Review Communication & Inspection Support: Coordinate communications with EMA or national authorities and provide guidance for QP inspections and GMP on-site audits.
5. Lifecycle Management: Post-approval variation submissions, PSUR (Periodic Safety Update Report) filings, and certificate renewal services.
For customized solutions, please contact us for professional support.
Registration Introduction1.Definition and Background
EU pharmaceutical product registration refers to the evaluation process by the European Medicines Agency (EMA) or national competent authorities to legally market drugs in the EU market. As a globally significant regulatory market, the EU registration system is renowned for its stringent requirements on quality, safety, and efficacy. For Chinese pharmaceutical companies, entering the EU market is not only a critical step in internationalization strategy but also an important approach to enhance production quality systems and global competitiveness.
2.Applicable Scope
New Drugs: Including innovative chemical drugs, biological products (e.g., monoclonal antibodies, recombinant proteins), and orphan drugs (rare disease medications).
Generic Drugs: Require demonstration of bioequivalence to reference drugs and are subject to decentralized procedures (DCP) or mutual recognition procedures (MRP).
Specific Categories of Drugs: Such as herbal medicines, modified-release drugs (e.g., extended-release formulations), and drugs already marketed in EU member states seeking market expansion.
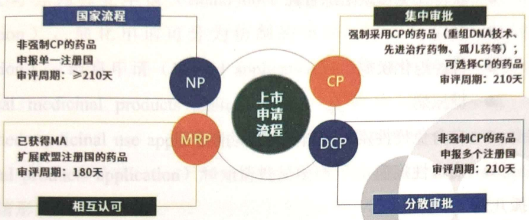
3.Basic Requirements
Qualification Requirements:
·MAH (Marketing Authorization Holder): Must be a registered company within the EU.
·MIAH (Manufacturing/Importation Authorization Holder): Must possess EU GMP certification.
·Quality management system, personnel qualifications (e.g., QP - Qualified Person), and facility compliance.
Technical Documentation:
·Complete CTD-format dossier covering pharmaceutical, non-clinical, and clinical data (where applicable).
Quality System:
·Compliance with EU GMP Annex requirements, including process validation, stability studies, and impurity control (e.g., mutagenic impurities per ICH M7).
4. Registration Process
Example: Decentralized Procedure (DCP)
DCP Submission Process:
1)Scheduling Submission (Identify RMS, book timeslot)
2)Pre-submission (Optional, at least 2 months before submission)
3)MAA Submission (via CESP or National Portal)
4)Validation (Technical + Regulatory)
5)Fee Payment (Proof of payment required before/after MAA submission)
DCP Approval Process:
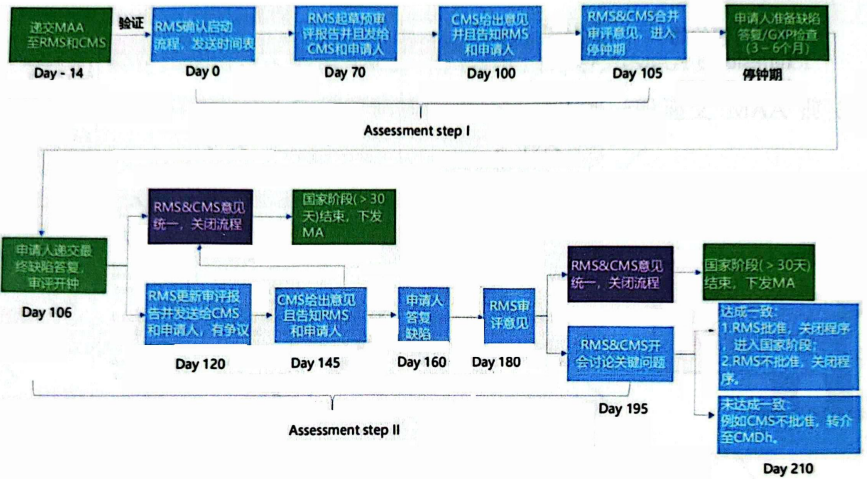
5.Regulatory Basis
·Directive 2001/83/EC: Regulates marketing authorization, production and distribution of human medicinal products.
·EU GMP Guidelines: Cover specific requirements for pharmaceutical quality management systems.
·ICH Guidelines: Including ICH Q11 (API development), ICH M7 (control of mutagenic impurities), etc.
·Special Product Regulations:
Biological products follow EMA biosimilar guidelines.
Herbal medicines refer to HMPC standards.
Product RecommendationLearn more about related businesses


Message Advisory
 Official account
Official account WeCom
WeCom- +86 (311) 6656 8233
- info@evalaboratories.com
- Chinese company address10th Floor, Building 1, Tech Centre, No.856, Zhongshan East Road, High-Tech Zone, Shijiazhuang, Hebei
- US company address813 Towne Center Drive, Pomona, CA 91767
©2025 Shijiazhuang Elab Biotech Co., Ltd. All Rights Reserved
冀ICP备2025127575号-1
冀ICP备2025127575号-1