+86(311)6656 8233
Services > Regulatory Registration > International Registration Services > DMF/VMF Registration (U.S. FDA)
DMF/VMF Registration (U.S. FDA)
 Our Services
Our Services1. DMF/VMF Documentation Preparation: Provide Drug Master File (DMF) and Veterinary Master File (VMF) application services for manufacturers of APIs, intermediates, packaging materials, excipients and other products.
2. Review Support: Services including Deficiency Responses (until FDA approval is obtained), DMF/VMF Supplements (for administrative and/or quality changes), Annual Reports, etc.
3. U.S. Agent Services: Professional compliance agency to ensure smooth communication channels.
For customized solutions, please contact us for professional support.
Registration Introduction1.Definition and Background
DMF (Drug Master File) is a confidential technical document required by the U.S. Food and Drug Administration (FDA). It is submitted by suppliers of pharmaceutical ingredients, excipients, packaging materials, or manufacturing processes to support regulatory applications such as New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs). Its primary purpose is to protect suppliers' proprietary information while allowing dosage form manufacturers to reference the DMF number directly in their submissions, thereby streamlining the review process and accelerating drug approval timelines.
VMF (Veterinary Master File): Similar to DMF but applicable to veterinary medicines, with essentially identical submission requirements and processes.
2.Applicable Registration Scope
U.S. DMFs are categorized into 4 types:
Type II: Drug substances, drug substance intermediates, materials used in their preparation, or drug products
Type III: Packaging materials
Type IV: Excipients, colorants, flavors, and their materials
Type V: FDA-accepted reference information
3.Basic Registration Requirements
Document completeness: Must include manufacturing process, quality control, stability studies, analytical method validation, etc., compiled in ICH M4 CTD format (electronic submissions must comply with eCTD standards)
U.S. agent: Non-U.S. companies are strongly advised to appoint a U.S. agent for FDA communications and document receipt
Compliance commitment: Must demonstrate compliance with cGMP (Current Good Manufacturing Practice) and submit annual updates (at minimum); significant changes require timely submission of amendments.
4.Registration Process
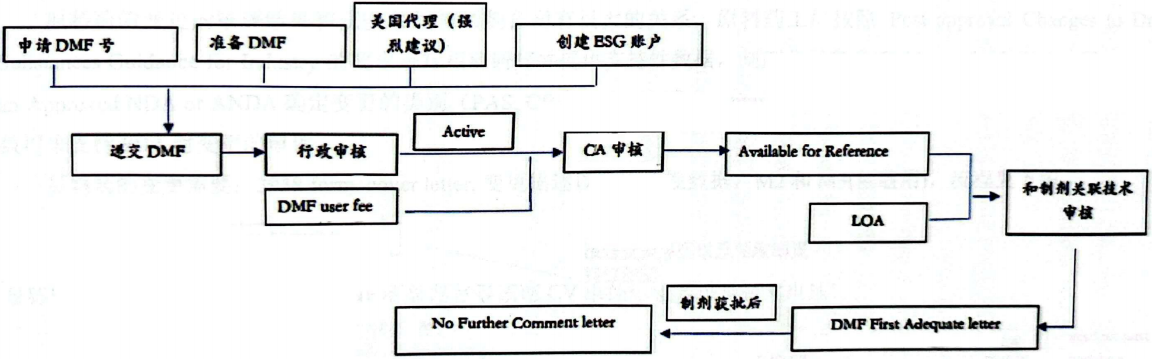
Note: Without reference from a drug product manufacturer, the DMF will only undergo a Completeness Assessment (CA) after fee payment, and will not proceed to technical review.
5.Regulatory Basis
·21 CFR 314.420: Legal framework for DMF submission and cross-referencing.
·ICH Q7: GMP standards for active pharmaceutical ingredients (APIs). DMF holders must commit to compliance with these guidelines.
·GDUFA (Generic Drug User Fee Amendments): Requirements for completeness assessment and fees for Type II DMFs.
FDA Guidance Documents:
·Guidance for Industry: Drug Master Files
·Container Closure Systems for Packaging Human Drugs and Biologics (for Type III DMFs)
Product RecommendationLearn more about related businesses


Message Advisory
 Official account
Official account WeCom
WeCom- +86 (311) 6656 8233
- info@evalaboratories.com
- Chinese company address10th Floor, Building 1, Tech Centre, No.856, Zhongshan East Road, High-Tech Zone, Shijiazhuang, Hebei
- US company address813 Towne Center Drive, Pomona, CA 91767
©2025 Shijiazhuang Elab Biotech Co., Ltd. All Rights Reserved
冀ICP备2025127575号-1
冀ICP备2025127575号-1